

HLRCC Handbook
Hereditary Leiomyomatosis and Renal Cell Cancer is a very rare genetic condition that was named in 2002. This handbook has been created to help educate and support all those impacted by HLRCC
Welcome Friend
Chances are that if you are reading this page, you or someone you know has been impacted by HLRCC. We are very glad that you are here; to gather information, learn about this condition, and most importantly, to take control of your own, your patient’s, or your loved one’s health.
If you want a quick overview of HLRCC, there is a 2 page hand-out called “QUICK FACTS,” to print and keep handy.
The term HLRCC did not exist a few years ago. Other older terms for this condition are Reed’s syndrome or MCUL. Then ten years ago the connection to the possibility of developing kidney cancer and the role of the FH gene was found giving rise to the new term. Communicating to the outside world about this condition has been slow and arduous, although we are making progress. So far, up to 1000 individuals have been involved in studies regarding this condition, but most likely thousands more individuals are going undiagnosed. In our attempt to protect ourselves and our family members, those of us diagnosed with HLRCC have learned the value of annual screening. If people with HLRCC are going to stay healthy, they have to understand the risks and take action to protect themselves. These facts, along with the knowledge that most doctors do not even know that this condition exists, prompted us to create this handbook.
The purpose of the HLRCC Foundation Handbook is:
- To teach patients how to take control of their own health once they are diagnosed
- To provide medical professionals with a convenient summary of the latest information available on HLRCC, and how best to manage the health of a patient with HLRCC.
- To communicate the importance of both screening for kidney tumors and conducting genetic testing for blood-related family members
- To offer doctors and patients resources for clinical research, tips for efficient and safe screening, and the most up-to-date information possible regarding current research and statistics for HLRCC
Finally, it is our hope that the HLRCC Foundation through its web site, Facebook Group, and this handbook will provide you with emotional support. An additional online resource is Inspire where those impacted by HLRCC, VHL, and BHD can exchange information, stories and encouraging words. We encourage you to join and partake. They are wonderful resources!
Thank you for your support and for doing all you can to spread the word about HLRCC. Remember, KNOWLEDGE SAVES.
HLRCC Foundation (Formerly HLRCC Family Alliance)
At the urging of Lindsay Middelton, Genetic Counselor at the US-NCI, and with the support of Joyce Graff of the then VHL Family Alliance, (Now VHL Alliance), Phil Thayer founded the HLRCCF in the autumn of 2004 and continued as leader of the group until early 2011. Key Milestones
In February 2011, Julie Haff Rejman became the Chair of the organization. Graham Lovitt became the Vice Chair.
In January 2013, Antony Horton Ph.D., also became a Vice-Chair.
In December 2022, Bruce Lee one of our Vice-Chairs and Driven To Cure CEO sadly died.
In March 2023, The rebranded HLRCC Foundation was defined.
HLRCC Foundation Leadership
Privacy Policy
The HLRCC Foundation website previously subscribed to the principles of HONcode of the international Health on Net Foundation, to assure you of the highest quality of health information.
Medical information on this site is reviewed by our Medical, Research and Support Council.
Information provided on the website is designed to support, not replace, the relationship that exists between a patient or site visitor and his or her physician.
The website does not accept advertising. It is supported solely by donations from people with HLRCC, their friends, their families and supporters, and physicians and researchers interested in HLRCC.

Privacy
Personal details that you provide to the HLRCC Family Alliance, including e-mail addresses, are kept entirely confidential. These details are shared within this organization among staff and volunteers for the purpose of providing service to you, but are never shared with, rented or sold to other organizations. All staff and volunteers have made confidentiality agreements to protect your information. To verify your information or send corrections, please contact us at hlrcc@vhl.org
Information you submit to us voluntarily through the website is stored on our secure server using SSL encryption technology.
Computer Tracking and Cookies
The website is not set up to track, collect or distribute personal information not entered by visitors. Our site logs do generate certain kinds of non-identifying site usage data, such as the number of hits and visits to our sites. This information is used for internal purposes by technical support staff to provide better services to the public and may also be provided to others, but again, the statistics contain no personal information and cannot be used to gather such information.
The website also recognizes the online site where a visitor searched to find a subject which brought them to the HLRCCFA website, but we cannot identify the visitor or the visitor’s address.

Site information is used to help us serve these search sites with the correct information about our material. No personal information is collected
A cookie is a small amount of data that is sent to your browser from a Web server and stored on your computer’s hard drive. HLRCCFA does not use cookies in its web pages. We do not generate personal data, do not read personal data from your machine and do not store any information other than what you voluntarily submit to us.
Problems or Complaints with HLRCCFA Privacy Policy

Links to Third Party Sites
The links included within the service may let you leave this site. The linked sites are not under the control of HLRCC Family Alliance and HLRCCFA is not responsible for the contents of any linked site, or any link contained in a linked site, or any changes or updates to such sites. These links are provided as a convenience only, and the inclusion of any link does not imply endorsement by HLRCCFA of the site or any association with their operators.
Medical, Research and Support Council
Current Members
Professor, Head of the Department of Clinical Genetics, HUSLAB ,Helsinki University Hospital, Finland
Dermatologist and Associate Editor of the Journal of the American Academy of Dermatology, Greenwood Village, CO, USA
Carlos Alberto Fredes
Executive Director of the Argentina Association of Families of Von Hippel-Lindau (VHL-AAF) Argentina
Programme Leader in Cancer Metabolism, MRC Cancer Unit University of Cambridge, UK
Research Group Leader, Beatson Institute of Cancer Research, Glasgow, Scotland, UK
Vice-Chair HLRCC Foundation, retired software quality assurance manager, software systems designer, Liverpool, UK
Professor of Medical Genetics and Genomic Medicine, University of Cambridge, UK
Consultant Clinical Geneticist, VU University, Amsterdam, The Netherlands
Professor of Oncogenetics and Chair, National Expert Centre for Rare Cancers PREDIR, Le Kremlin-Bicêtre, France
Laura S. Schmidt, Ph.D.
Urologic Oncology Branch, National Cancer Institute*, USA
Pamela Stratton, M.D.
Gynecologist, National Institute of Child Health and Human Development, US-NIH,Bethesda, MD, USA
Honorary Members
Although no longer actively involved, in recognition of their contribution to the world of HLRCC the following have kindly agreed to be listed.
Virpi Launonen
Finland, Author of the paper establishing the connection between symptoms of HLRCC and the gene on chromosome 1 (2001)
Phil Thayer
USA, Founder of HLRCC Family Alliance in 2004
Supporting the HLRCC Foundation c/o VHLA
The original HLRCC Family Alliance was founded in 2004 as a support group for people affected by Hereditary Leiomyomatosis and Renal Cell Cancer and interested health care professionals, and to promote research. Funding the VHL Alliance helps support our website and any outreach we do to the medical community, including such things as domestic and international mailings, attending medical conferences related to HLRCC, or supporting travel expenses for HLRCC educators. The VHL Alliance is funded by the generosity of its supporters. The HLRCC Fouudation is currently not a separate legal entity and is kindly supported by VHL Alliance infrastructure. It is intended for the HLRCC Foundation to become a non-profit charity with the tax authorities in the United States.
* The VHL Alliance is registered as a non-profit charity with the tax authorities in the United States, Canada, and Great Britain, as well as other countries. Please contact your local group and your tax advisor for specific information on guidelines for tax deductibility of donations.
Please mail the following Supporter/Donation form to HLRCC Foundationc/o VHL Alliance, 1208 VFW Parkway // Suite 303, Boston, MA 02132 USA. Thank you!
Supporter/Donation Form
QUICK FACTS
- HLRCC stands for Hereditary Leiomyomatosis and Renal Cell Cancer (sometimes Carcinoma). It is also known as Reed’s Syndrome.
- HLRCC is caused by an inherited genetic alteration in the Fumarate Hydratase (FH) gene. There is a 50% risk of passing this on and the severity of the disease can vary a lot from person to person. It can be diagnosed by the detection of this genetic alteration (mutation).
- Many women with HLRCC develop large uterine fibroids in their twenties. Although benign, the fibroids may result in early treatment.
- Both men and women tend to develop benign skin leiomyomas (or “skin bumps”) in their twenties. These two symptoms together, fibroids and skin leiomyomas, offer an important clue to the need for genetic testing of the FH gene. Because of the absence of uterine fibroids, HLRCC is more likely to go undetected in men, and early diagnosis is less likely.
- Screening requires an annual MRI scan of the abdomen using preand post- contrast 3D acquisition with 3mm slice reconstruction for the detection of kidney cancer. Be sure to read the Handbook section as well.
- “Suggested Screening Guidelines” for more information.
- Even small HLRCC kidney tumors can metastasize, or spread, very quickly to the bones, lungs and brain. Unlike some other cancers, there is no curative treatment for kidney cancer once it metastasizes, although life may be extended with the latest class of drugs. Annual screening helps our goal to prevent metastasis and to stay healthy.
- If you live in the United States, you may want to consider being part of a clinical trial at the National Institutes of Health (US-NIH) in Bethesda, MD. This is the only trial open at the time of writing. We expect that more will be opened in the future, possibly at other locations around the world.
- It is recommended that children who have a parent with HLRCC have genetic testing by age 8 as screening is recommended from that age for children at risk. Please read more about this in the Handbook.
- It is a newly identified condition (2002) and is currently being studied at several locations around the world. There are approximately 200-300 families currently diagnosed with HLRCC with perhaps 1500 patients. It is an under-diagnosed condition because of its rarity.
- Being diagnosed with HLRCC can be a very scary thing, mostly because it has the word cancer in its heading. If you have HLRCC it does not mean that you have kidney cancer or will necessarily get it. However it does mean that you have an increased risk factor for kidney cancer, and you need to screen yourself so that doctors can detect even the smallest HLRCC cancer in your kidneys.
- Knowledge truly is a gift.
AN OVERVIEW: What is HLRCC?
In recent years, scientists have used the work of the Genome Project to help identify new connections between physical symptoms that used to be viewed as isolated or random. One of these diseases is called HLRCC, or Hereditary Leiomyomatosis and Renal Cell Cancer. HLRCC is a rare inherited condition first fully described in 2001. It is caused by a tiny alteration in one copy of the FH gene.
According to researchers at the US-NIH HLRCC is a rare inherited condition characterized by the presence of cutaneous leiomyomas, uterine fibroids, and/or kidney cancer. A person who is diagnosed with HLRCC has inherited a susceptibility to develop one or more of these symptoms.
Approximately 450 people have been evaluated at the US-NIH, along with many other people in England, France, Japan, Finland, and Australia, but it is believed that there are many more people who are undiagnosed. Considering HLRCC is a relatively new, and rare condition, there remains much to be learned.
There is considerable variation in symptoms from family to family and among members of the same family. Each person in a family has their own individual susceptibility to the symptoms. For example, if your parent had kidney cancer, it does not mean you will develop a kidney tumor. You have your own unique susceptibility.
Even if a child inherits the altered gene, it does not necessarily mean that he or she will have any symptoms of HLRCC. There may never be any symptoms at all. Or that person may develop just one of the issues, or two, or possibly all three. So far, researchers have not been able to find any patterns that would allow one to predict which symptoms a person will develop based on the particular genotype they have. However, there do seem to be trends in some families. Some families seem to only get leiomyomatosis and other families are more likely to get RCC (kidney cancer).
HLRCC is caused by having an alteration (mutation) in one copy of the Fumarate Hydratase (FH) gene. The FH gene is a section of DNA that codes for a protein called fumarase. Fumarase is an important enzyme needed for the production of energy by mitochondria, which are the tiny organelles inside our cells that produce most of our cells’ energy. All people have fumarase and researchers are trying to learn the normal function of fumarase and why alterations in the FH gene cause the symptoms of HLRCC.
HLRCC is inherited in an Autosomal Dominant manner, meaning that both males and females can be affected, and each child of an affected parent has a 50% chance of inheriting the altered FH gene.
All people have two copies of the Fumarate Hydratase gene, one from their mother and one from their father. In a person with the condition called HLRCC, one of their two genes is normal and working well. The other is altered, meaning that it has a change in it and does not work very well. The altered FH gene is unable to produce the fumarase enzyme properly. When a person has only one working copy of the gene, their cells make less fumarase than normal, but enough to be healthy. During a person’s lifetime, genes can become damaged when alterations occur during cell division or from exposure to chemicals or radiation. Cells have the ability to repair damaged DNA, but sometimes they cannot, leaving a non-working altered gene in a cell. In a person with HLRCC, an alteration in the second copy of the FH gene can lead to biochemical changes that cause those cells to grow into benign smooth muscle tumors (leiomyomas) of the skin and uterus, or less often malignant tumors of the kidney. For more detailed information regarding the FH enzyme, please refer to our HLRCC Science.
* The VHL Alliance is registered as a non-profit charity with the tax authorities in the United States, Canada, and Great Britain, as well as other countries. Please contact your local group and your tax advisor for specific information on guidelines for tax deductibility of donations.
Please mail the following Supporter/Donation form to HLRCC Foundationc/o VHL Alliance, 1208 VFW Parkway // Suite 303, Boston, MA 02132 USA. Thank you!
Diagnostic Criteria
Although there is no consensus on diagnostic criteria, the US-NIH and other experts believe that an individual has HLRCC if they have any of the major features of the condition, including:
Cutaneous leiomyomas: Most individuals with HLRCC present with multiple cutaneous leiomyomas (skin bumps). A single cutaneous leiomyoma on its own is insufficient to confirm an HLRCC diagnosis unless another family member has already an HLRCC diagnosis. However, continuing to be vigilant and possibly having a single baseline MRI scan of the kidneys should be followed.
Uterine Leiomyomas: Most females with HLRCC have uterine fibroids, often quite large and occurring early in their 20s. However, fibroids are very common in the general population and are rarely diagnostically useful on their own.
Kidney (Renal) tumors: Most people who have HLRCC do not develop kidney tumors. The incidence of kidney tumors in the US-NIH study group is about 30%. However, the incidence of kidney tumors in the European group is reported as much lower. We are still learning the specific factors that increase or decrease one’s risk of kidney cancer.
A positive genetic test for HLRCC: This means that an alteration has been detected in the Fumarate Hydratase gene. FH alterations can be found in about 97% of families who are strongly suspected of having HLRCC.
If HLRCC is suspected, but the genetic alteration cannot be found and there are no cutaneous leiomyomas, then a fumarase enzyme assay can be done on cells derived from skin or blood. A fumarase activity level less than or equal to 60% is indicative of HLRCC. This test is specialized and it is not available in most laboratories. Some laboratories that can test for fumarase activity find difficulties because of problems in calculating and interpreting the results.
Recently some pheochromocytomas and paragangliomas have been found to be associated with HLRCC and there is now a new section added to this Handbook.
There are other cancers which have been occasionally associated with HLRCC. Breast and prostate are examples. Some HLRCC family members have other health problems (an example is thyroid nodules), but it is not clear if these are related to HLRCC. At this point they are assumed to be coincidental. We are all still members of the general population.
Note: Researchers in England discovered a difference between HLRCC tumors and non-HLRCC tumors (kidney and other tumors). This is called the immunohistochemistry for 2-Succinocysteine (2SC) test. Fumarase deficient cells give positive staining meaning that the cells are HLRCC positive.
Note: There are other tumor types where the number of cases is too small to allow us to categorize these tumors as diagnostic criteria for HLRCC, but when they occur in an HLRCC patient, the tumors are found to have no fumarase activity. Examples are benign adrenal tumors and Leydig testicular cancer which develops in the Leydig cells — the cells in the testes that release the male hormone, testosterone. There is also a possibility of benign ovarian cystadenomas and some Wilms’ tumors being associated with HLRCC.
Although there is no consensus on diagnostic criteria, the US-NIH and other experts believe that an individual has HLRCC if they have any of the major features of the condition, including:
Cutaneous leiomyomas: Most individuals with HLRCC present with multiple cutaneous leiomyomas (skin bumps).
Uterine Leiomyomas: Most females with HLRCC have uterine fibroids, often quite large and occurring early in their 20s. However, fibroids are very common in the general population and are rarely diagnostically useful on their own.
Kidney (Renal) tumors: Most people who have HLRCC do not develop kidney tumors. The incidence of kidney tumors in the US-NIH study group is about 30%. However, the incidence of kidney tumors in the European group is reported as much lower. We are still learning the specific factors that increase or decrease one’s risk of kidney cancer.
A positive genetic test for HLRCC: This means that an alteration has been detected in the Fumarate Hydratase gene. FH alterations can be found in about 97% of families who are strongly suspected of having HLRCC.
If HLRCC is suspected, but the genetic alteration cannot be found and there are no cutaneous leiomyomas, then a fumarase enzyme assay can be done on cells derived from skin or blood. A fumarase activity level less than or equal to 60% is indicative of HLRCC. This test is specialized and it is not available in most laboratories. Some laboratories that can test for fumarase activity find difficulties because of problems in calculating and interpreting the results.
Recently some pheochromocytomas and paragangliomas have been found to be associated with HLRCC and there is now a new section added to this Handbook.
There are other cancers which have been occasionally associated with HLRCC. Breast and prostate are examples. Some HLRCC family members have other health problems (an example is thyroid nodules), but it is not clear if these are related to HLRCC. At this point they are assumed to be coincidental. We are all still members of the general population.
Note: Researchers in England have discovered a chemical compound that is present in HLRCC tumors, but not in non HLRCC tumors (kidney and other tumors), so in the future we may see newer and better screening tests for HLRCC.
Note: There are other tumor types where the number of cases is too small to allow us to categorize these tumors as diagnostic criteria for HLRCC, but when they occur in an HLRCC patient, the tumors are found to have no fumarase activity. Examples are benign adrenal tumors and Leydig testicular cancer which develops in the Leydig cells — the cells in the testes that release the male hormone, testosterone. There is also a possibility of benign ovarian cystadenomas and some Wilms’ tumors being associated with HLRCC.
* The VHL Alliance is registered as a non-profit charity with the tax authorities in the United States, Canada, and Great Britain, as well as other countries. Please contact your local group and your tax advisor for specific information on guidelines for tax deductibility of donations.
Please mail the following Supporter/Donation form to HLRCC Foundationc/o VHL Alliance, 1208 VFW Parkway // Suite 303, Boston, MA 02132 USA. Thank you!
Genetic Testing
HLRCC is an autosomal dominant disorder. “Autosomal” means that the alteration is located on one of the 22 regular chromosomes and not on a sex chromosome (X or Y). “Dominant” means that having just one copy of the altered gene is enough to cause the disorder. This means that if a person has HLRCC, there is a 50% chance that he or she will pass the altered gene to a child. There is a 50% chance that an embryo of an HLRCC parent will have the condition, depending on whether the particular egg or sperm from which that the embryo was formed contained the altered copy of the gene. You have two FH genes – one from each parent. The one healthy parent gives you one healthy unaltered FH gene. The other parent with the altered gene gives you one of their two copies of the gene: either a healthy unaltered FH gene or an altered FH gene – hence the 50% chance. You either have an altered gene or you don’t. Occasionally a person with an altered FH gene may have very few symptoms, so that it may seem to skip a generation, but if you do not have an altered FH gene you cannot pass it to a child. It is possible for an alteration in the FH gene to be present for the first time in one family member as a result of a mutation in a germ cell (egg or sperm) of one of the parents or in the fertilized egg itself. This is termed a “de novo mutation”.

Parent 1 with HLRCC
A = FH Gene Mutation
B = FH Working Gene

Parent 2 without HLRCC
C = FH Working Gene
D = FH Working Gene
Each child gets one copy of the FH gene from each parent.
In this way, there are four possible arrangements of these four genes. Each arrangement has a 25% chance.
So for each child there is a 50% chance of having HLRCC and a 50% chance of not having it.
AC: Child With HLRCC
A=FH Gene Mutation
C=FH Working Gene
AD:Child With HLRCC
A=FH Gene Mutation
D=FH Working Gene
BC: Child Without HLRCC
B=FH Working Gene
C=FH Working Gene
BD: Child Without HLRCC
B=FH Working Gene
D=FH Working Gene
Genetic testing is the most reliable way of diagnosing HLRCC. Although a physician can often diagnose an individual with HLRCC based on the physical signs listed above, the US-NIH recommend that individuals obtain genetic testing to confirm that they actually have the gene alteration.
Benefits of genetic testing to confirm HLRCC:
- A positive test (meaning you do have HLRCC) will help people advocate with their doctors and insurance companies for agreement with their need for annual kidney screening.
- It gives family members who do not have symptoms the ability to discover whether they have the gene alteration by a simple blood test.
- A negative test (meaning you do not have HLRCC) can put your own fears to rest when other family members have tested positive (meaning they have HLRCC).
- A negative test (meaning you do not have HLRCC) can put your own fears to rest when other family members have tested positive (meaning they have HLRCC).
- As we learn more, the exact alteration in your gene will become increasingly important to your own health maintenance.
Things to keep in mind regarding genetic testing:
- It may complicate your ability to obtain life or health insurance. Refer to section about Life and Health insurance. Genetic testing and lifetime kidney surveillance is expensive. It is much easier if a person has health insurance in place before obtaining a DNA test.
- It can be an emotional process. It is not a simple blood test. The results may be difficult to interpret, and it is best to undertake genetic testing through a genetic professional (geneticist or genetic counselor) who can help you understand the results and their implications for yourself and your family.
- If you have your children tested before their age of consent and they are found positive there are implications for future life or health insurance, and mortgage applications as well as the start of a life-long screening process. This is a difficult decision to make between health safety and financial implications. We recommend that people have life insurance in place even for children before obtaining a DNA test. Many countries are passing anti-discrimination laws to protect citizens.
If you are in the worldwide HLRCC clinical study at the US-NIH (Bethesda, Maryland, USA):
- Genetic testing at US-NIH is optional, so you can still be in the study and not have the genetic testing done. This is especially beneficial if you have physical signs and family members who have had positive genetic testing, but don’t want the testing done yourself. All testing at US-NIH is optional.
If you are not in the US-NIH study, you should look for a genetic counselor from your area online. The best links to use are:
US – https://www.nsgc.org/
World – https://geneclinics.org and go to the link for Clinic Directory
You will want to bring a copy of a clinical paper about HLRCC such as the Toro article https://www.ncbi.nlm.nih.gov/books/NBK1252/ with you to your genetic counselor and your genetic counselor is welcome to contact Lindsay Middelton at US-NIH or another expert consultant to obtain further information. You should also give your genetic counselor the link www.hlrccinfo.org so that they can make sure you get the screening you need when they connect you with the proper physicians, should your test be positive.
References:
Pithukpakorn M, Toro JR., Hereditary Leiomyomatosis and Renal Cell Cancer. GeneReviews [Internet]. 2010. PMID: 20301430,
https://www.ncbi.nlm.nih.gov/pubmed/20301430
https://www.ncbi.nlm.nih.gov/books/NBK1252/
The risk of developing the features of HLRCC increases with age. You are at significant risk of being diagnosed with an FH gene alteration if you have a diagnosed blood relative. The actual risk figure depends on the closeness of the relationship starting as high as 50% with a first degree relative (parent, child or sibling). There is a lower risk of 25% with a second degree relative (uncle/aunt, niece/nephew or a grandparent) and a still lower risk with a third degree relative (first cousin) or even a first cousin’s child. The alteration cannot however skip generations so the more genetic testing a family has the more precise the risk figure will be for each individual ranging from 0% to 50%. In other words, if one of your parents is at risk, but tests negative, then you will not be at risk. If a person does not carry the altered gene, they cannot pass it to a child. You may wish to discuss this with your genetic counselor for more clarification.
If you test positive and want to inform other family members, there is at the end of the handbook, a printable Family Letter (to send to relatives of someone recently diagnosed with HLRCC). The letter should only come from the person with the HLRCC diagnosis, or the next-of-kin if the person has died, in order to respect their privacy. It is important to keep in mind that your family members may be overwhelmed when they receive this information. We tried to keep this in mind when creating our printable letter and to offer the support of the HLRCC Foundation as a way of supporting each and every person associated with this condition.
If your genetic test is negative, but you have symptoms of HLRCC you should check that your genetic test covered the possibility of FH deletions including whole gene deletion, in addition to bidirectional sequencing. This is usually through a technique called MLPA or multiplex ligation probe amplification. Some companies only offer sequencing.
It can sometimes take several months to obtain genetic testing results, but once one genetic alteration is identified within the family, testing of additional family members is faster and less costly. Tests in some countries are faster than others. Please consider the following PROS and CONS when considering genetic testing as well:
PROS:
- Knowing about the HLRCC genetic alteration gives you the insight to screen and protect yourself or your siblings/children or other family members, if they are tested. Knowledge is power!
- Catching HLRCC tumors early may save your life. A kidney tumor often grows with no symptoms. Periodic screening with scans will catch a tumor early, so that it can be treated.
CONS:
- A positive test result may have an impact on your own and your children’s ability to get life or health insurance. You should be sure to have coverage before requesting DNA testing.
- The knowledge of a condition such as this has emotional implications. People can become very worried about the future, which is where a support group such as HLRCC Foundation www.hlrccinfo.org can help to put your life back on track.
Life and Health Insurance
A useful link describing the general implications for insurance (applies to all genetic conditions not just to Birt-Hogg-Dubé) ishttps://www.bhdsyndrome.org/for-families/additional-resources/insurance/ and there is information for different US States in https://www.healthinsuranceinfo.net/
Concerns about Genetic Discrimination of Health Insurers
Many people who learn they may have HLRCC are concerned that their health insurance company will either terminate their policy or deny coverage if the insurer learns of their genetic status. Many states have enacted state laws to protect their citizens from genetic discrimination by health insurers. However, the protection offered varies widely among state laws. You can access information about your state law by accessing: https://genome.gov > Issues in Genetics > Statute and Legislation Database https://www.genome.gov/PolicyEthics/LegDatabase/pubsearch.cfm
In 2008 the Genetic Information Nondiscrimination Act (GINA) was signed into law. The GINA Act prohibits discrimination by health insurance companies and employers based on “genetic information”, including information about genetic testing or your results (and those of your family members) or information about family history of any disease or disorder.
Health insurance protection:
- Group and individual health insurers may not use your genetic information to set eligibility, premium or contribution amounts;
- Health insurers may not request or require that you take a genetic test.
Employment protection:
- Employers may not use your genetic information to make decisions involving hiring, firing, job assignments and promotions;
- Employers may not request, require or purchase genetic information about you or one of your family members. GINA stipulates that genetic information alone cannot be considered a pre-existing condition.
What does GINA NOT do?
- GINA does NOT restrict health care providers from requesting, offering or providing information about a genetic test to patients.
- GINA does NOT require insurance companies to pay for any particular test.
- If you have been diagnosed with a medical problem or a symptom related to HLRCC, GINA does NOT apply
Employers with fewer than 15 employees and the military are not required to abide by the employment protections.
Having Children
Deciding whether to have children when there is a 50% chance of inheriting a problem is a difficult decision to make. Before making any decision you may wish to speak with a geneticist or genetic counselor about possible testing options and its implications. One option may be Pre-implantation Genetic Diagnosis (PGD). There is information on the VHL Alliance website about PGD. See https://www.vhl.org/
Other options may include Chorionic Villus Sampling (CVS) or amniocentesis.
It’s advisable for the other non-HLRCC parent to be genetically tested for any FH Gene variant that is known to be pathological for Fumarase Deficiency to avoid the possibility of having a Fumarase Deficient child.
Cutaneous Leiomyomas (Skin Bumps)
There is considerable variation in the appearance of cutaneous leiomyomas as can be seen in the following photographs making it difficult to diagnose by sight unless you are a specialist.
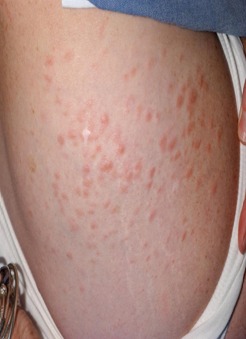
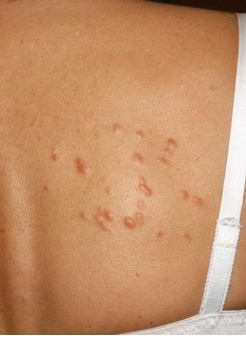
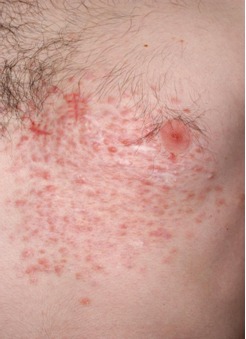
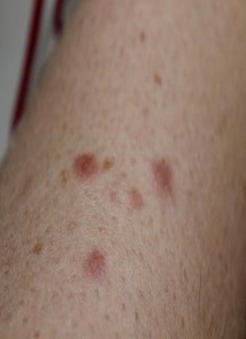
Photos 2, 3 and 4 courtesy of Dr. Ed Cowen, NIH, NCI.
Just as uterine leiomyomas grow from smooth muscle of the uterus, cutaneous leiomyomas are rare benign tumors that grow from smooth muscles in the skin. The arrectores pilorum (singular arrector pili, also called piloerectus muscles) are small smooth muscles that are attached to hair follicles. These are the muscles that allow your hairs to “stand up” when you are cold or fearful. When benign tumors grow from arrrectores pilorum, they are called piloleiomyomas. They also may be called cutaneous leiomyomas. It is estimated that most people with HLRCC will get one or more piloleiomyomas in their lifetime.
Piloleiomyomas are very helpful for identifying people who are likely to have HLRCC.
Individuals who have HLRCC tend to have cutaneous leiomyomas by the time they are in their 30’s, but some have gotten them as early as 11 years old; some people report finding their first one in their 50’s.They tend to grow anywhere on the body and limbs, but rarely on the face, hands or feet. These bumps can be very small, but sometimes large, can range in color from skin-colored to light brown to red, and tend to grow in mosaic clusters, but can also be solitary. Some people have a single leiomyoma, but many people develop small clusters of leiomyomas. A smaller percentage of people develop wide distribution of leiomyomas over their chests or backs (“segmental distribution”). Once a leiomyoma appears, it does not go away.
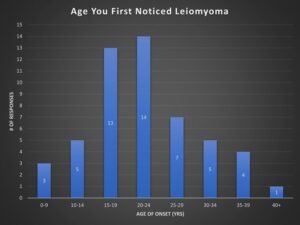
A survey taken in the HLRCC Foundation’s Facebook Group shows the onset age of cutaneous leiomyoma in a sample size of 53 members.
Leiomyomas can be mistaken for post-acne scarring. They are very firm to the touch and discrete as compared to acne or eczema. They are not common skin lesions in the general population, but dermatologists are starting to recognize them more easily as potential indicators of HLRCC.
Diagnosis
A skin biopsy must be done to confirm a leiomyoma, as relying on appearance is inconclusive. A skin biopsy involves minor surgical removal of some of the skin bump, after which the tissue is sent to a pathology lab. An anesthetic agent is injected under the skin around the leiomyoma and once the area is numb, a small sample of tissue is taken. Slides are prepared from the tissue and examined under a microscope by a pathologist to determine whether the diagnosis is piloleiomyoma or some other type of growth. This is a technical description: The smooth muscle fibers are fusiform and are composed of eosinophilic cytoplasm with elongated blunt-ended (spindle shaped) nuclei and perinuclear halos in cross-sections. Cutaneous leiomyomas are clinically very similar to other lesions and require a biopsy and histologic evaluation for confirmation. The clinical differential diagnosis includes nevus, dermatofibroma, neurofibroma, glomus tumor, angiolipoma, and other benign growths, hamartomas or neoplastic lesions. The immunohistochemistry for 2-Succinocysteine (2SC) test can be used on the removed tissue to give a precise diagnosis.
Cutaneous leiomyomas can be the only clue to a physician about whether a person is at risk for HLRCC. If one has a first-degree relative with HLRCC, then a single confirmed cutaneous leiomyoma is sufficient for the diagnosis of HLRCC. Without a first-degree relative with HLRCC at least two are required for a HLRCC diagnosis.
Pain
As with all the conditions associated with HLRCC there is considerable variation not only in the appearance of cutaneous leiomyomas, but also in the experience of pain in them. The exact cause of pain has not been understood, but there is a thought that the leiomyoma has trapped nerve cells. The variation is not just from patient to patient, but also within one patient and can increase over time. Some patients find a cold sensitivity to such an extent that they even consider moving to a warmer country. Some find that if a pain develops in one leiomyoma it acts as a trigger to all the others to become also painful for hours or days at a time. Sometimes a leiomyoma that grows initially without having any pain symptom can start to become irritable and painful. Pain can sometimes occur as a result of exertion, or by touch or rubbing clothes. When pain does occur most patients describe it as excruciating, like having a knifepoint stab.
Treatment
Cutaneous leiomyomas are often sensitive to cold and touch, but many people do not find them painful. However, in some people leiomyomas are very painful. Most dermatologists do NOT recommend surgically removing all these growths, except as needed for biopsies, as it tends to cause scarring and divots in the skin where the procedure is performed. However painful or unsightly leiomyomas, if there are not too many close together, can be surgically removed using CO2 laser or freezing with cryosurgery. Preferably the removal is by a skilled plastic surgeon to minimize scarring. Use of a super-fine thread avoids the dots on either side of the line of surgery. Removal will normally be done under a local anesthetic. There is a reported case of extensive multiple piloleiomyomas being successfully removed by surgery and reconstructed with a flap technique. Sometimes the leiomyomas will grow back after removal, possibly because some tissue was left behind, or there were new ones growing when in a cluster. You should talk with a dermatologist about what is best for your type of skin growth.
Some of our members have reported (2021) improvement by taking Vitamin D3. The maximum recommended dose is 4,000 i.u. and to help absorption of Vitamin D take with Vitamin K2 100μg (available in combined form). As always you should consult with your medical practitioner.
Researchers at US-NIH experimented with Botox therapy to help with the pain (see Clinical Trials) but there was no improvement. If you have painful cutaneous leiomyomas, you may want to get a contact name from info@hlrccinfo.org or talk with your own dermatologist about options.
One of our members has found significant relief using Lyrica. As with any treatment you should first discuss and agree its suitability with your physician and or dermatologist. The links below have a lot of information about Lyrica including descriptions of warnings and side effects which seem important to study before deciding to take it.
https://www.lyrica.com/PHN/phn-introduction.aspx and https://arthritis.about.com/od/pregabalin/a/Lyrica.htm and https://en.wikipedia.org/wiki/Pregabalin
Another pain relief drug that is being used is niphediprine. As with the use of all drugs you should consult with your medical team – especially so if intending to become pregnant.
Significant pain relief has also been reported with pulsed hysocine butyl bromide see https://www.ncbi.nlm.nih.gov/pubmed/20049277. This article mentions many calcium channel blockers like nifedipine, phenoxybenzamine, doxazocine, gabapentin and topical 9% hyoscine hydrobromide.
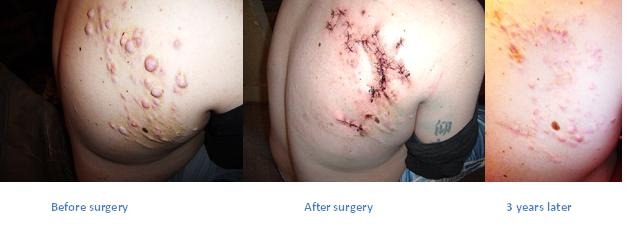
Courtesy of Ryan N, HLRCC patient – showing significant growing back after 3 years.
A dermatologist should conduct an annual inspection of all cutaneous leiomyomas to detect changes which might lead to malignant cutaneous leiomyosarcoma (which is a rare cancer, but may be 2% in HLRCC see paper “Reassessing the clinical spectrum associated with hereditary leiomyomatosis and renal cell carcinoma syndrome in French FH mutation carriers”).
There has been one reported case of a solitary angioleiomyoma with multiple piloleiomyomas see https://www.ncbi.nlm.nih.gov/pubmed/10356411.
Uterine Fibroids
Like the skin, the uterus contains smooth muscle tissue, and uterine fibroids are smooth muscle tumors that grow in the wall of the uterus or womb,(myometrium) and with the ligament of the womb. They are almost always benign (non-cancerous) and can be as small as an apple seed or as large as a grapefruit. Other medical terms for fibroids are myoma fibromyoma leiofibromyoma fibroleiomyoma and fibroma (the dual of myoma is myomas or myomata).
In the general population, up to 80% of women develop uterine fibroids by the age of 50 years. Uterine fibroids are the leading reason for hysterectomies in the United States (1 in 3 have fibroids). Women with fibroids may be at greater risk of having a cesarean section when they give birth. Other women with fibroids may have difficulty becoming pregnant or carrying a pregnancy. More often fibroids are simply “innocent bystanders” during pregnancy and cause no problems at all.
Uterine fibroids are often the first physical symptom to develop in a female who has HLRCC. Although a nuisance, they provide a clue to individuals who are at risk, as well as a warning to be looking for skin leiomyomas, which are often the second physical symptoms of HLRCC.
The growth of uterine fibroids is believed to be affected by hormones (byespecially the female hormones estrogen and progesterone). The occurrence of fibroids is genetic. Uterine fibroids that are associated with HLRCC tend to be larger and to occur at earlier ages than in the general population.
If you are planning to have children, it may be better to have them in your 20’s rather than to wait until your 30’s, as over time fibroids may begin to create issues which can complicate fertility or pregnancy.
Fibroid Symptoms
Fibroids are so common – and can be so small – that many women do not even know that they have them. Women with HLRCC often have moremore and much larger fibroids that are accompanied by other symptoms, including:
- Heavy bleeding
- A feeling of fullness in the pelvic area
- Enlargement of the abdomen
- Frequent urination
- Pain during sex
- Lower back pain
- Complications during pregnancy and labor
Diagnosis
Ways to find out if you have uterine fibroids:
- A gynecology (GYN) pelvic examination (a doctor can often detect larger fibroids just by feeling the abdomen).
- An ultrasound scan
- Hysterosalpingogram – where dye is injected into the uterus
A pelvic ultrasound scan is usually performed transvaginally. However, when the uterus is very large, a transabdominal pelvic ultrasound may be needed to measure the full size of the fibroids and uterus. Some radiologists and gynecologists also perform a sonohysterogram in which a catheter is placed in the cervix into the uterus and fluid (sterile water or saline) is infused. Sonohysterograms can be helpful in determining whether there are fibroids within the uterine cavity.
Hysterosalpingogram is an x-ray test done during testing for infertility, and is used to investigate the shape of the uterine cavity and whether the fallopian tubes may be blocked. During hysterosalpingograms, radio-opaque material is injected into the uterine cavity and x-rays are done. Hysterosalpingograms can be helpful in determining whether there are fibroids within the uterine cavity or whether these fibroids compress the opening to the fallopian tubes. However they are not useful in looking at the fibroids themselves, and not all centers perform this procedure.
A gynecologist should continue these annual checks and consider adding radiological examination. Once a woman with HLRCC is known to have fibroids, a gynecologist should perform an annual examination to check for increasing uterine size suggestive of fibroid growth. Having imaging studies done over time (ultrasound or MRI) will also enable the radiologist and gynecologist to determine whether the fibroids have grown in size or number. Significant growth in fibroid size might suggest the development of a malignant type of fibroids called leiomyosarcoma. Thus, when fibroids grow rapidly, surgical treatment is usually recommended. Hysterosonograms and hysterosalpingograms may be helpful as a preoperative assessment for myomectomy (described below). While the first reports of HLRCC suggested that leiomyosarcoma might occur in women with fibroids, a large study at US-NIH has shown that fibroids may be atypical, but no cases of leiomyosarcoma have been observed.

From https://en.wikipedia.org/wiki/Uterine_fibroid
A moving story: “I was at the University of Michigan as a high-risk patient while I was pregnant with my first child,” said Julie Sherwood, a 39-year-old HLRCC patient. “I had a vertical c-section and they still could not find my baby because I had so many fibroids in the way. They had to do a second incision and the baby was saved. During my second pregnancy my fibroids tripled within weeks and the baby couldn’t survive. I had an emergency hysterectomy weeks later.”
Julie’s moving story, along with many other discussions about a variety of health-related topics, can be found in a radio interview at:
https://powerfulpatient.org/archive/2011/week1111_fibroids.php
Treatment
There are several options of managing fibroids. Many factors are considered to determine which option is the best one for each individual.
Sometimes a hysterectomy is recommended. During hysterectomy the uterus and cervix are removed, and sometimes the ovaries are removed as well. It is not possible to carry a pregnancy after the uterus is removed.
Another option for treating fibroids is a surgical procedure called myomectomy. During a myomectomy only the fibroids are removed, and thus the ability to carry a pregnancy may be preserved. When fibroids are within the uterine cavity, they may be removed through a hysteroscopic myomectomy procedure (operating using a viewing instrument through the cervix). When the fibroids are large and within the wall of the uterus, an abdominal myomectomy is usually performed. While some gynecologists perform this procedure laparoscopically or robotically (operating using a viewing instrument in the abdomen), it is important that the fibroids are not morcellated (mechanically cut up into pieces) inside your body. Removing the fibroid intact will enable the pathologist to examine it for atypical features or possible leiomyosarcoma. Additionally, there is a concern that many fibroids associated with HLRCC are atypical and cellular. With morcellation, small pieces of tissue left in your body may attach and grow. Another concern is the number of myomectomy procedures done prior to attempting pregnancy. If a woman undergoes several myomectomy procedures prior to attempting pregnancy, she may have difficulty becoming pregnant or may be at risk of miscarriage. These effects may occur because, for example, of scarring in the uterine wall. Thus, timing myomectomy surgery just before attempting pregnancy may be a preferable strategy.
Non-surgical treatments of uterine fibroids include Uterine Artery Embolization (UAE) or High Intensity Focused Ultrasound Ablation (HIFU) of fibroids. Both procedures are performed by an interventional radiologist. These physicians are trained in a medical sub-specialty of radiology which utilizes minimally-invasive image-guided procedures to treat diseases. At the present time, neither procedure is recommended for women with HLRCC as they will not investigate whether fibroids are atypical or cancerous. These strategies either lessen the flow of blood to the uterus (UAE) or physically destroy part of the fibroid (HIFU). In some patients with HLRCC who have undergone UAE, their fibroids have rapidly increased in size.
Birth control pills and the levonorgestrel (MirenaTM) IUD decrease menstrual bleeding in women regardless of whether they have fibroids, and are effective methods of contraception. Women with the HLRCC gene defect are able to use these treatments for these indications. While fibroids do appear to be hormone-sensitive, it is unknown at this time whether using hormones like birth control pills or the levonorgestrel IUD will be effective in decreasing fibroid size in women with HLRCC.
There are many organizations and links that will assist in your understanding of uterine fibroids. Keep in mind that we are currently trying to educate these organizations about HLRCC and that you most likely will not find information specific to HLRCC on these websites.
The Center for Uterine Fibroids
https://www.fibroids.net
The National Uterine Fibroids Foundation
Colorado Springs, Colorado
(719)633-3454
https://www.huff.org
The following links provide additional information about fibroids:
Renal (Kidney) Tumors
Note: This section of the Handbook is also relevant for some non-HLRCC tumors which are Fumarate Hydratase Deficient (FHdRCC).
Research indicates that only a proportion of individuals with HLRCC develop malignant (cancerous) kidney tumors. There is a higher proportion in the US-NIH study possibly because they are recruiting as kidney cancer specialists. The variation in incidence figures is considerable and may depend on several factors including methods of recruitment. It will take more studies to determine the true risk of kidney tumors in a particular individual. Meanwhile, since this is such a potentially lethal tumor, we recommend that everyone be screened for kidney tumors. See Suggested Screening Guidelines.
Diagnosis
Kidney cancer often develops with no initial symptoms, and by the time symptoms such as pain or blood in the urine appear, it has often already become a danger to life.
In the general population kidney cancer accounts for about 1-2% of all cancers. Renal Cell Carcinoma(RCC) is divided into groups : Clear cell, Papillary, Oncocytic and Chromophobe, Collecting Duct, Other Renal, Molecularly Defined. In Molecularly Defined there is Fumarate Hydratase-deficient renal cell carcinoma which includes RCC associated with HLRCC and also sporadic cases.
A person with a diagnosis of HLRCC has a much higher risk of developing lesions in the kidney compared to the general population. There are three kinds of lesions in the kidney: benign cysts, hard tumors, and hard lumps developing inside a cyst. Unlike other genetic syndromes associated with kidney cancer, HLRCC kidney tumors can metastasize (spread) when the tumor is relatively small (less than 1 cm). These tumors usually spread to the lungs, bone and brain. It was thought that HLRCC was generally unilateral in that only one kidney is initially affected. However as people are living longer after surgery, problems can develop in the remaining kidney. Like all kidney tumors they often initially have few, if any, noticeable symptoms, which is why scanning is essential in order to detect them at an earlier stage.
Although a kidney biopsy is now considered safe in the general population it should be avoided in HLRCC patients to avoid the possibility of spreading cancer cells. Instead, a diagnosis from MRI and CT scans is recommended by an HLRCC expert.
Treatment / Management of the Kidneys
This is an area which is still being actively researched and there are many factors to be considered for every individual. The key to managing HLRCC kidney tumors is surveillance to find tumors when they are small. Surgical removal if possible is currently the treatment of choice when a tumor is found. Some families report a currently healthy grandparent who had a nephrectomy many years ago, and there are people who had tumors removed 10 years ago who are healthy and well.
Once an HLRCC kidney tumor has metastasized, the prognosis changes, and the course of metastatic kidney cancer is similar to the experience of others in the general population. There are targeted molecular treatments that are currently in clinical trials. At the US-NIH there is a clinical trial of a combination of bevacizumab (Avastin) and erlotinib (Tarceva). In 2014 the initial report of the trial was very favorable. The spectrum of available drugs and the recommendations for primary and secondary therapies for papillary kidney cancer is evolving rapidly. Check with an oncologist experienced in kidney cancer and make contact with an HLRCC specialist with advice from info@hlrccinfo.org.
The type of kidney surgery is dependent on many factors. Sometimes it may be necessary to remove the entire kidney (nephrectomy) or a partial nephrectomy may be performed. A partial nephrectomy is nephron sparing surgery in which only the tumor and a little surrounding margin of tissue is removed. With HLRCC a wider margin should be used.
Follow-up after RCC
The exact follow-up procedure for every patient will vary from center to center and should be discussed and agreed with your physicians. We will only describe here examples of what you may expect.
If you have had a nephrectomy, then initially for the first 2 years a full chest and abdominal CT scan with contrast may be carried out every 6 months to check for any metastasis (spread). ALWAYS TELL THE RADIOLOGIST THAT YOU HAVE ONLY ONE KIDNEY AND PLEASE TO REDUCE THE AMOUNT OF CONTRAST.
A PET/CT and a bone scan may also be ordered if metastasis is thought likely. Kidney cancer will often metastasize to the lungs or bone. Note this is then called secondary kidney cancer in the lung or bone and NOT lung cancer or bone cancer.
After two years the screening interval may be increased to yearly, but there continues to be a life-time risk of either recurrence or metastasis requiring long-term surveillance. See https://www.ncbi.nlm.nih.gov/pubmed/21903243
Pheochromocytomas and Paragangliomas
Recently ~2013 it has been discovered that some pheochromocytomas and paragangliomas are related to Fumarase Hydratase (FH) Gene – HLRCC .
By kind permission from the VHL Alliance, this comprehensive section is copied from its Handbook. Although the section is with reference to VHL it can be regarded as also relevant to HLRCC. Most HLRCC diagnosed people will never develop a pheochromocytomas (PCC) or a paraganglioma (PGL) in their lifetimes but the possibility exists and patients and physicians should be aware of their symptoms.

Catecholamines: This is predominantly epinephrine, but also some norepinephrine. Epinephrine helps to regulate the “fight or flight” response to stress (also known as adrenaline. It is the main catecholamine produced by the adrenal glands).
Glucocorticoids: The most important glucocorticoid is cortisol. Cortisol helps to regulate blood sugar, blood pressure, fat and protein metabolism, and the immune system. Cortisol is known as the ‘stress hormone.’
Mineralocorticoids: The most important mineralocorticoid is aldosterone. Aldosterone works mainly in the kidneys by maintaining salt and water balance within the body. This is important for blood pressure regulation and proper cardiovascular function.
Adrenal androgens: These are precursors to sex hormones (i.e. testosterone, estrogen).
Pheochromocytomas and Paragangliomas
VHL is associated with a usually benign type of tumor occurring in the adrenal glands, called a pheochromocytoma (pheo). Pheos occur more frequently in some families than others. They are rarely malignant (less than 7% of the time) among people with VHL. Detected early, pheos do not cause problems, but they are potentially lethal if not treated. This is especially true during times of heightened stress (surgery, accidents, childbirth, etc.).
Pheos that develop outside of the adrenal glands are called paragangliomas (paras) and are very rare, even in VHL patients. Paragangliomas may occur anywhere on the sympathetic nervous system, which includes anywhere along a
line drawn from the groin to the ear lobe, on either side of the body. Multiple tests may be needed to find them.
Research indicates that adrenal tumors are as much as four times more common among people with VHL than previously thought. Even in families that have not previously had a pheo, it is still important to test for the presence of these tumors. In one large progeny in France where there were no pheos for three generations; there are now pheos in two branches of that family.
Symptoms
The primary clinical sign of a pheo is high blood pressure, especially spiking blood pressure, that can put strain on the heart and vascular system, potentially causing a heart attack or stroke. However, blood pressube normal, despite the presence of a pheo. Patients may notice headaches, irregular or rapid heartbeat, or what feels like a panic attack, fear, anxiety, or even rage. There may be unexplained heavy sweating and sometimes people experience hot and/or cold flashes. There may also be abdominal pain or unexplained weight loss. It is recommended that all people with VHL be screened regularly for pheos.
Testing for a Pheochromocytoma
It is extremely important to test for pheochromocytomas before undergoing surgery for any reason, as well as before going through childbirth. Undergoing either of these stressful experiences with an undiagnosed pheo can be extremely dangerous. If the doctors are aware that the pheo is there, they can take preventive action that will ensure the safety of the patient and of any unborn child.
Traditional blood or urine tests that measure only catecholamines are inadequate to find most pheos. In order to diagnose a pheo, an initial biochemical test is done to measure blood or urine metanephrines. The preferred test is a
plasma free metanephrines test, due to its higher sensitivity. This involves taking a sample of blood and measuring the levels of metanephrine, the metabolite of adrenaline, and normetanephrine, the metabolite of noradrenaline. More widely available is the fractionated 24–hour urinary free metanephrines test, which includes collecting a 24–hour urine sample and analyzing it for fractionated metanephrines and normetanephrine.
The measurement of normetanephrine is most important, since VHL–related pheos usually do not produce adrenaline or its metabolite, metanephrine, in significant amounts. Testing for methoxytyramine (a metabolite of dopamine) levels may be useful in evaluating metastatic status, although only 17% of VHL patients with pheos produce it. If additional information is required, or if there are symptoms of a pheo, but the blood and urine tests are negative, anatomical imaging scans should be used.
Testing Standards for Pheochromocytomas and Paragangliomas
Below are the clinical guidelines that were approved by the Endocrine Society for testing for the presence of pheochromocytomas and paragangliomas (together known as PPGLs):
Surveillance for PPGLs should always include measurements of plasma metanephrines (obtained from a blood sample) or urinary fractionated metanephrines (obtained from a urine sample).
For a blood sample, it is recommended that patients be supine (lying down) for a minimum of 20–30 minutes between the time the needle is inserted and the time the blood is drawn. Studies have shown that seated blood testing more often results in false positives. The reason for this is that the release of catecholamines by peripheral nerves and the adrenal gland is stimulated by an upright posture. Sitting upright results in increased blood levels of metanephrines compared to being in a supine position.
For blood sample analysis, upper reference intervals (the test result above which a pheo is determined to be present) should be established from supine tests, not seated tests, in order to minimize the chance of a false negative result (missing a PPGL that is present).
Analysis is performed using liquid chromatography (LC–MS/MS) with mass spectrometric, or electrochemical detection and using supine norms for plasma test results. All positive test results should be followed up. Follow–up may involve repeated biochemical studies (e.g. a clonidine test), or a CT scan or MRI (if a CT scan is not appropriate).
In VHL, it is only necessary to consider elevations of normetanephrine. For plasma in an adult patient with VHL, anything over 112 picograms/milliliter (0.61 nanomoles/liter, the NIH upper reference limit) should evoke suspicion. Anything over 400 pg/mL (2.2 nmol/L) for a sample that is taken with the patient lying down
and relaxed (no stress) and on no antidepressants is immediately highly suspicious (close to 100% likelihood). Imaging is then warranted. Between those ranges, the likelihood of a pheo increases with increased level and follow–up tests, such as imaging, should be considered.
If these chemical tests indicate the presence of a pheo, but it cannot easily be located on MRI or CT, an MIBG or PET scan may be recommended. These scans help to locate a pheo, even if it is outside the adrenal glands.
According to research at the U.S. National Institutes of Health, different tests
have different success rates in locating a pheochromocytoma or paraganglioma:
- 18F–FDA PET scan finds 75–92%
- 18F–FDOPA PET scan finds 67–93%
- 123I–MIBG scan finds 67–86%
- 18F–FDG PET scan finds 83–93% (adrenal: 67%)
- Octreotide scan finds fewer than 50% of these tumors. Please note that the Octreotide scan will soon be replaced by 68Ga–DOTA analogs used with PET scans.
The choice of one of these tests is often made depending on the availability of a particular technology at that center. However, it is important to note that if the test chosen does not find the pheo, there is still some chance that the pheo is there but cannot be detected by that particular test. A second opinion should be requested from a VHL or pheochromocytoma expert.
Preparing for Pheochromocytoma Testing
The accuracy of the urine and blood tests for pheo activity will be determined in large part by your own cooperation in preparing for the test. The tests are most reliable when care is taken in two areas—diet prior to testing and preservation of the urine sample from the start of the test until the lab processing is complete.
For a reliable measurement of plasma and urinary free metanephrines, no specific diet is required. Testing for plasma or urinary 3–methoxytyramine requires a diet poor in catecholamines to prevent false–positive results. Catecholamines can be found in fruits (such as bananas), fruit drinks, and nuts.
While there is no strong data that suggests that regular use of tobacco or alcohol may be linked to inaccurate results, caffeine should be avoided because it can cause false–positives on some tests. Certain drugs and medications can interfere with the measurement method being used, while others, like antidepressants, can cause false–positives. Be sure to tell your doctor and the technician if you are taking any medications. If possible, testing for pheos should be done before beginning any medications.
Specific instructions may differ slightly from center to center, due to different methods of analysis. Follow any instructions carefully to avoid a false reading.
Preparation for Blood Testing
The procedure usually takes about 45 minutes. It is important that you be quiet and calm for 20–30 minutes prior to the blood draw to ensure accurate results. Bring something with you to keep you occupied and relaxed as you will be
asked to lie quietly on a table for 20 minutes after the needle is inserted before
the test begins.
Table 4. Published upper limits for reference intervals of plasma concentrations of metanephrines in children (from samples collected lying down with an indwelling i.v.).
Reference intervals for each lab may be slightly different due to variations in processing.
If there are concerns about interactions with medications, it is important that the laboratory use LC–MS/MS techniques to analyze the sample, in order to achieve the highest sensitivity and selectivity in checking fractionated
Preparation for 24–hour Urine Testing
Pro Tip: Do not begin collection on Friday or Saturday. This ensures that your sample will be delivered to the lab on a working day and can be processed promptly.
1. Start the collection in the morning. Empty the bladder; do not save this urine specimen.
2. Write the date and time on the jug. (If there is a preservative added to the jug, be careful not to get it on the skin. If this happens, wash the area immediately with water.)
3. Save all the urine passed for the next 24 hours in the jug provided including the final specimen passed exactly 24 hours after beginning the collection.
4. Keep the urine refrigerated at all times. You might keep it in a paper bag in the refrigerator. If you must be out, you could carry it in a bag or backpack with plastic ice packs against the jug.
5. Write this date and time on the jug when the collection is finished.
6. Bring the collection and the paperwork to the lab as soon as possible after collection. (Labs are usually open early in the morning or have a place where you can arrange to drop it off early.)
Treatment
If surgery is required, the preferred method is a cortical–sparing partial adrenalectomy. Studies have shown that keeping even a small amount of the cortex of the adrenal gland, if surgery on both glands is required, makes it easier to manage post–surgery. It also usually avoids the need for steroid replacement.
On the other hand, it must also be recognized that the remaining adrenal tissue can be associated with recurring pheos. Removal of the entire adrenal gland is rarely required to manage VHL–associated pheos.
The “key hole” operating technique (laparoscopy) is currently used to treat pheos. With this technique, there is less risk of infection and the recovery is much faster. Some surgeons have the technology to simultaneously remove pheos located on each of the two adrenal glands. Laparoscopic or robotic surgery should be discussed with your doctor.
Prior to surgery, the medical team will prescribe “blockers” (alpha blockers, sometimes followed by beta blockers), or drugs that inhibit the formation of catecholamines. These medications will calm the effects of the chemicals produced by the pheo and allow the surgery to proceed calmly, without causing a pheo crisis. While the blockers will make you tired, they are critically important.
They may be prescribed for two or more weeks before the planned surgery.
Another important consideration before surgery is to make sure that the anesthesiologist working with your surgeon has experience with pheos. The anesthesiologist is responsible for managing your blood pressure during the surgery. Your endocrine surgeon should be able to let you know who will be part of your surgical team.
Adrenal Dietary and Lifestyle Management Strategies
Maintain a healthy diet. Chronic stress is associated with increased levels of cortisol, a hormone related to stress which helps regulate blood sugar, blood pressure, fat and protein metabolism, and the immune system. High levels of cortisol can promote overeating and lead to weight gain. Eating a balanced and nutritious diet supplies the body with all its essential nutrients and can be useful for controlling weight, reducing stress, and improving performance.
A clinical study evaluating the effect of calorie restriction for one month in
otherwise healthy overweight women aged 20–36 found that, along with an
average weight loss of almost 13 pounds, there was a significant decrease in blood
pressure, heart rate, and cortisol hormone levels, improved hand–eye
coordination, and no evidence of increased physiological or psychological stress.
Work with a dietician to develop the most appropriate diet for you.
Eat salt and stay hydrated. Those who have had their adrenal glands removed due to pheochromocytomas or who have adrenal insufficiency (or Addison’s Disease) generally need more salt in their diet. This is because they do not have enough of a hormone called aldosterone, which regulates sodium and potassium (salt and electrolyte) levels in the body. Aldosterone is produced by the adrenal glands, and without functioning adrenal glands, there is very little or no aldosterone production. If aldosterone levels get too low, the body loses too much sodium.
People who produce low or no amount of aldosterone are often categorized as “salt–wasters” because they cannot maintain salt (sodium) levels. These individuals must take supplements to replace aldosterone. Even with replacement, maintaining optimal levels of aldosterone can be a challenge. When these “salt wasters” exert themselves heavily or spend enough time in hot temperatures, there is a good possibility of their losing too much salt in sweat and urine, putting them at a higher than normal risk for dehydration. Therefore, “salt wasters” should be sure to drink enough non–sugar–loaded liquids and supplement with enough salt to alleviate this dangerous situation. Good liquid options include water (always the best choice), seltzer or soda water, tea of any type, fruit juice, milk, broth, etc.
Avoid simple carbohydrates. Cortisol is released by the adrenal glands if the body has low blood sugar. Low levels of glucose can occur when meals are skipped or taken at irregular intervals. Eating simple or refined carbohydrates (such as sugar, corn or table syrup, or white flour) can also cause low blood sugar levels, since they are digested and absorbed very quickly by the body. Instead of a gradual rise in blood sugar, this quick absorption triggers a quick spike in blood sugar levels that is followed by a quick decline. This rapid increase and decrease in blood sugar levels causes an increase in cortisol levels, which triggers the stress response mechanism.
Eating meals at regular intervals and consuming foods other than simple carbohydrates can prevent this increase in cortisol levels. Proper diet is important not only to control blood sugar and reduce spikes in stress hormone levels, but also to reduce risk factors for disease.
Limit stimulants. Consumption of stimulants, such as energy drinks, coffee,
or soft drinks has been linked to feeling stressed. The effect of caffeine is known to increase cortisol hormone production and intensify the stress response.
Therefore, caffeine should be consumed in moderation or avoided by people exposed to chronic stress or with impaired adrenal function. Smoking cigarettes can also increase stress; nicotine exposure is known to increase cortisol levels.
Stay positive, practice self–care. Low self–esteem and loneliness are known to increase cortisol levels, while maintaining a positive outlook on life and a good social support system is associated with lower stress hormone levels.
Sleep. There is a known association between sleep and levels of cortisol, the stress hormone. While getting enough quality restful sleep can slightly decrease cortisol levels, disturbed sleep or not getting enough sleep can lead to mildly increased cortisol levels. For this reason, sleep deprivation may be an important risk factor leading to stress–related disorders.
Medication. Please note that if you have had both of your adrenal glands completely removed, it is important to follow your prescribed daily doses of hydrocortisone and fludrocortisone, and to be checked regularly by your endocrinologist. These medications work to replace the functions of your missing adrenal glands to manage the balance of fluids in your body, maintain kidney function, control blood pressure, and maintain cardiovascular health.
TYPES OF SCANS
There are four types of Kidney Imaging: Magnetic Resonance Imaging (MRI), Computed Tomography (CT), Positron Emission Tomography (PET) and Ultrasound. Some centers will alternate MRI with CT scans. The kidney cancer of HLRCC can metastasize (spread to other organs or bone) even when small, which makes early detection essential. There are several well-used methods to visualize kidney tumors. There are pros and cons for each.
Magnetic Resonance Imaging (MRI)
MRI scans with and without contrast is an acceptable method to examine kidneys. This process creates images using magnetic waves. No radiation is used.
The MRI scan for HLRCC patients should use pre-and post-contrast 3D acquisition with as high a resolution as possible, 3mm or less slice reconstruction is advised. Clinicians at the National Institutes of Health are currently using 1mm slice reconstruction in this population. The scans should be reviewed by someone with expert HLRCC knowledge especially if kidneys cysts are present. The preferred option is to use MRI with gadolinium contrast, because it is the contrast medium that identifies soft tissue. Please make sure that specific contrast agents are used (macrocyclic agents, not linear agents). Gadobutrol is one example. See Extracellular Gd agents . There is some concern that repeated use of gadolinium contrast may build up in the body tissue and some people have serious side effects from the contrast agent. It may be possible to get sufficient quality images without using gadolinium contrast. MRI scanners have recently improved with “Open MRI” and shorter and wider tunnels making the experience less problematic even for people with mild claustrophobia. However, there is some concern that the image quality may be reduced with some open large bore MRI scanners. The objective is to get the best possible picture quality. If you feel you need calming medication for anxiety or discomfort, a larger bore to accommodate larger body mass, or any other requests, negotiate a solution that will give the doctor the picture quality needed while addressing your concerns. Ask: Is it possible to enter the MRI machine feet first? People find this is an improvement on the unpleasant experience. Note: Breast tissue expanders with magnetic ports are MRI unsafe, preventing patients from benefiting from the diagnostic capabilities of MRI.
Computed Tomography (CT Scan)
CT scans with and without contrast is an acceptable method of visualizing kidneys. However, images are created through the use of radiation. Annual CT scans over a life span raises concerns about using up your lifetime maximum of radiation exposure.
The CT scan is done with a contrast agent usually containing iodine and you should drink plenty of water afterwards to flush the contrast agent out of your body. If there is any doubt about your kidney function (creatinine levels above 1.6) you may be given 500cc of saline solution first and Visipaque(iodixanol) or Omnipaque (iohexol) contrast which have lower iodine content. The unpleasant, oral, barium-based contrast used to distinguish the digestive organs has been phased out in the U.K. and replaced with just drinking plain water. Radiologists in the U.K. said it was just as good.
Computed Tomography Photon-Counting
This photon-counting CT Scan is using new technology (2021) at NCI/NIH to give improved scans with less radiation see pubs.rsna.org/doi/10.1148/radiol.2018172656 for a description of it.
Positron Emission Tomography (PET)
For ordinary RCC, PET scans are normally less effective than CT scans with contrast and have been known to give false negatives. A PET scan is often combined with a CT scan. However, with HLRCC any tumors present are glucose hungry and PET can be a useful diagnostic tool especially for detecting metastases. There can be false positives in both the adrenal glands and the uterus because of the presence of benign HLRCC related tissue. Cancer cells upregulate glucose metabolism, which is a phenomenon known as the Warburg effect. This is the basis for PET in which a glucose analog tracer FDG (2-18fluoro-2-deoxy-D-glucose), a radioactive modified hexokinase substrate, is used to differentiate between normal and tumor tissue.
You may be offered a “delayed PET scan” see Advantage of delayed whole-body FDG-PET imaging for tumour detection.
CLINICAL TRIALS
The list of clinical trials and studies is constantly changing and people are advised to seek out the latest information from your medical specialists or by contacting info@hlrccinfo.org. The following was current at the time of writing this document.
The Natural History Study at the National Institutes of Health
The US-NIH, currently has a Natural History study for individuals who have been diagnosed with HLRCC or who have the clinical symptoms that might imply a possible diagnosis.
If you have an interest in the US-NIH Study, contact:
First point of contact:
Debbie Nielsen, BSN
Genetic Counselor
Urologic Oncology Branch
National Cancer Institute
National Institutes of Health
Bldg. 10, CRC, Rm. 2W-5740
9000 Rockville Pike
Bethesda MD 20892
T: (240) 760-6247
F: (301) 480-2869
Email: Deborah.Nielsen@nih.gov
Debbie will then send details for scheduling to the patient coordinator
Brittany Dawson-Wade (C) currently on extended leave 3rd March, 3023
Replacement Stefania Misul see email below
Patient Care Coordinator III
Clinical Monitoring Research Program Directorate (CMRPD)
Frederick National Laboratory for Cancer Research
Leidos Biomedical Research, Inc.
Support to: Urologic Oncology Branch
National Institutes of Health
10 Center Drive, Room 2-5750
Bethesda, MD, 20892
T: (240) 529-8590
[Email: Brittany.Dawson@nih.gov]
For HLRCC drug trials, please contact
Erin N. Purcell, BSN, RN
Research Nurse Specialist
National Cancer Institute
National Institutes of Health
9000 Rockville Pike
Bethesda MD 20892
T: (240) 858-3933
F: (301) 480-2869
Email: erin.purcell@nih.gov
The following was current at the time of writing this document.
The Natural History Study at the National Institutes of Health
This study started in 2002 is still currently recruiting participants. The study is now seeing new people with HLRCC who have a kidney cyst or a solid tumor and also one new first person from a new family with HLRCC. Other people at risk in HLRCC families should be screened by their local medical centers. The Trial Number was NCT00055627(now obsolete).
UCLA HLRCC Registry and Study
UCLA has created a registry to characterize the burden of HLRCC, determine who is at risk for skin, uterine, adrenal, and kidney manifestations, and help find new cures for this condition. Patients at other centers have successfully had their tumors sent in to make new models of HLRCC kidney cancer. Please contact them at kidneycancer@mednet.ucla.edu
Phase II Pamiparib and Temozolomide
https://clinicaltrials.gov/show/NCT04603365
“A Phase II Study of Pamiparib and Temozolomide for the Treatment of Hereditary Leiomyomatosis and Renal Cell Cancer”
Brief Summary:
Estimated start date: October 24, 2021
This phase II trial investigates how well pamiparib and temozolomide work in treating patients with hereditary leiomyomatosis and renal cell (kidney) cancer. Poly adenosine diphosphate-ribose polymerase (PARPs) are proteins that help repair DNA mutations. PARP inhibitors, such as pamiparib, can keep PARP from working, so tumor cells can’t repair themselves, and they may stop growing. Chemotherapy drugs, such as temozolomide, work in different ways to stop the growth of tumor cells, either by killing the cells, by stopping them from dividing, or by stopping them from spreading. Giving pamiparib and temozolomide may help treat patients with hereditary leiomyomatosis and renal cell cancer.
Collaborators:
BeiGene www.beigene.com/
Driven To Cure www.driventocure.org/
Phase II Study of Bevacizumab and Erlotinib
http://clinicaltrials.gov/show/NCT01130519
“A Phase II Study of Bevacizumab and Erlotinib in Subjects With Advanced Hereditary Leiomyomatosis and Renal Cell Cancer (HLRCC) or Sporadic Papillary Renal Cell Cancer”
This clinical trial of bevacizumab (AVASTIN® | Genentech, Inc.) and erlotinib (Tarceva® is ongoing, and is active but not recruiting patients with papillary kidney cancer that has spread (metastasized) beyond the kidneys. Very good interim results have been reported in http://medicalxpress.com/news/2014-11-kidney-cancer-patients-combination-anti-cancer.html Kidney cancer patients respond well to a combination of two existing anti-cancer drugs
In Annual Meeting News W. Marston Linehan, M.D is reported as saying:
“As for HLRCC syndrome, NCI began focusing on this familial kidney cancer syndrome in the 1980s. Researchers learned that these patients develop a particularly aggressive form of type 2 papillary kidney cancer. In addition to raised and painful cutaneous leiomyomas, 90 percent of female patients in these HLRCC syndrome families also develop uterine fibroids.”
“HLRCC is one of the most malignant types of kidney cancer there is,” Dr. Linehan said. “It needs to be detected because it can spread early and can be lethal.”
HLRCC syndrome is caused by an alteration of the Krebs cycle enzyme, fumarate hydratase. In studying this fumarate hydratase pathway in HLRCC, NCI researchers learned that when the fumarate hydratase gene in the cancer cell is damaged, it alters its metabolism significantly, becoming exceptionally dependent on glycolysis and glucose uptake.
“We have developed an approach to treatment that involves using bevacizumab and erlotinib, therapeutic agents that target the vulnerability of this fumarate hydratase pathway in HLRCC patients with advanced kidney cancer,” Dr. Linehan said. “This is currently an ongoing clinical trial, and we are cautiously optimistic about the early results.”
He also reported that blood tests are available to assist clinicians in making a diagnosis by detecting fumarate hydratase (FH) for HLRCC syndrome and folliculin (FLCN) for BHD syndrome.
“In both of these hereditary cancer syndromes, it is important for dermatologists and other clinicians to understand the significance of fibrofolliculomas and leiomyomas,” Dr. Linehan said. “These patients need to be evaluated for the possible presence of kidney cancer when they present with these dermatologic findings.” Listen to W. Marston Linehan, M.D (MP3)
Editor’s Note: Additional drugs will be forthcoming that will target the FH pathway. Stay tuned for more news
Phase I/II Study of Vandetanib and Metformin
https://clinicaltrials.gov/ct2/show/NCT02495103
“Vandetanib in Combination With Metformin in People With HLRCC or SDH-Associated Kidney Cancer or Sporadic Papillary Renal Cell Carcinoma”
Terminated (Vandetanib is no longer available as Sanofi has decided not to provide additional drug. All subjects are off-study.)
This Phase I/II clinical trial of vandetanib (CAPRELSA® | AstraZeneca United States ) and metforminwas for patients with papillary kidney cancer that has spread (metastasized) beyond the kidneys
Phase II Trial of the DNA Methyl Transferase Inhibitor Guadecitabine (SGI-110)
https://clinicaltrials.gov/ct2/show/NCT03165721
A Phase II Trial of the DNA Methyl Transferase Inhibitor, Guadecitabine (SGI-110), in Children and Adults With Wild Type GIST,Pheochromocytoma and Paraganglioma Associated With Succinate Dehydrogenase Deficiency and HLRCC-associated Kidney Cancer
Wild-type gastrointestinal stromal tumor (GIST) is a cancer in the esophagus, stomach, or intestines. It does not respond well to standard chemotherapy or radiation therapy. Most people with GIST are treated with imatinib. But it may not work in many children with GIST. Researchers think the drug SGI-110 may help treat people with GIST, pheochromocytoma and paraganglioma (PHEO/PGL), or kidney cancer related to hereditary leiomyomatosis and renal cell carcinoma (HLRCC).
Phase I Study of the Glutaminase Inhibitor CB-839 in Solid Tumors
https://clinicaltrials.gov/ct2/show/NCT02071862
This phase I trial using CB-839 is recruiting (December, 2015) patients with Fumarate Hydratase (FH)-deficient tumors.
Phase 2 Trial Olaparib with and without AZD1775, AZD5363, and AZD2014 in Treating Patients with Advanced Solid Tumors
https://clinicaltrials.gov/ct2/show/NCT02576444
Researchers at Yale have just opened a clinical trial that they believe specifically addresses the underlying biology of HLRCC. (They are actively looking for HLRCC patients at this time). Their description in lay language: Cancer patients with a family history of FH or SDH mutations, or tumors found to have these mutations. These mutations most typically appear in patients with hereditary kidney cancers including syndromes such as HLRCC. Genetic testing must have been completed that confirms the presence of this mutation. For patients with a history of HLRCC, the investigators at Yale can assist with confirmation of the mutation in the tumor(s). HOW do I get more information? Contact ranjit.bindra@yale.edu or brian.shuch@yale.edu
There is more information in a conversation in Smart Patients.
Treatment of Cutaneous Leiomyomas with Botulinum Toxin
http://clinicaltrials.gov/show/NCT00971620 “Randomized Pilot Study for the Treatment of Cutaneous Leiomyomas with Botulinum Toxin”
This study is completed and NIH has published a result “The use of botulinum toxin to treat painful cutaneous leiomyomas was associated with improved quality of life and with a trend toward improved pain at rest.” Efficacy of Intralesional Botulinum Toxin A for Treatment of Painful Cutaneous Leiomyomas: A Randomized Clinical Trial.
Intravenous Recombinant Human IL-15
http://www.clinicaltrials.gov/ct2/show/NCT01021059
“A Phase I Study of Intravenous Recombinant Human IL-15 in Adults With Refractory Metastatic Malignant Melanoma and Metastatic Renal Cell Cancer”
This study has completed. No results yet published.